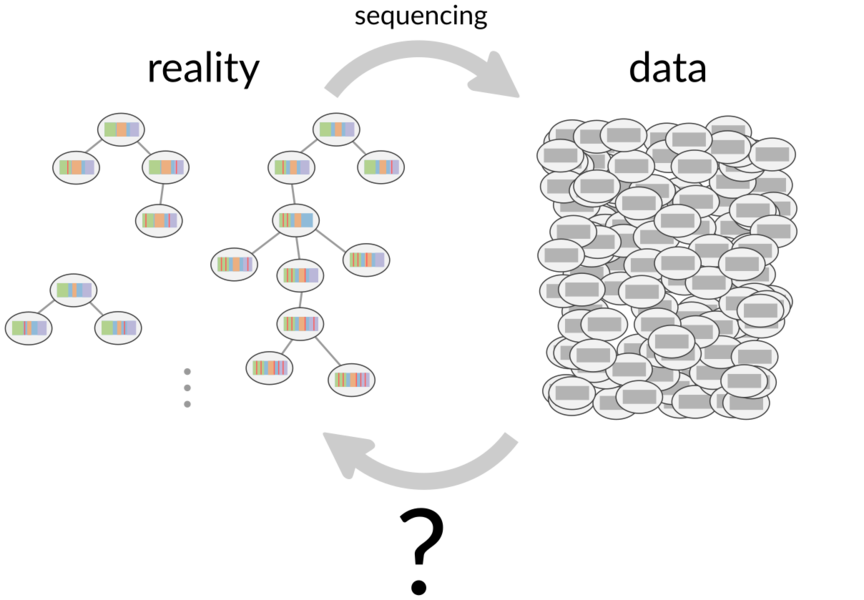
 Antibodies are encoded by B cell receptor (BCR) sequences, which (simplifying somewhat) arise via a two-stage process.
The first stage is a random recombination process creating a so-called naive B cell, which are the common ancestors in the trees to the right.
The second is initiated when such a naive cell (perhaps weakly) binds an antigen, and consists of a mutation and selection process to improve the binding of the BCR to the antigen.
The history of this process can be thought of as a phylogenetic tree descending from these naive common ancestors.
One can sequence the B cell receptors resulting from these processes in high throughput, which form an implicit record of these complex processes in a single individual.
Antibodies are encoded by B cell receptor (BCR) sequences, which (simplifying somewhat) arise via a two-stage process.
The first stage is a random recombination process creating a so-called naive B cell, which are the common ancestors in the trees to the right.
The second is initiated when such a naive cell (perhaps weakly) binds an antigen, and consists of a mutation and selection process to improve the binding of the BCR to the antigen.
The history of this process can be thought of as a phylogenetic tree descending from these naive common ancestors.
One can sequence the B cell receptors resulting from these processes in high throughput, which form an implicit record of these complex processes in a single individual.
The situation from a phylogenetic inference perspective is a mess. Sampled sequences have typically been mutated substantially from their ancestral naive sequence. Thus given a pair of sequences sampled from the repertoire, it’s not clear if they share a common ancestral naive sequence— that is, if they even belong in the same tree. Furthermore, in healthy individuals the size of these trees is very small compared to the number of sequences in the total repertoire. This makes for a difficult, and very interesting, clustering problem.
Duncan Ralph and I have been working on this problem since he arrived several years ago, and I am very happy to announce that we’ve put up a manuscript describing that work on arXiv. This paper builds on our previous work on BCR sequence annotation and alignment using a hidden Markov model. Using this HMM, we can define a likelihood for observing a given set of sequences distributed among a given collection of clusters. This likelihood integrates over possible alternative annotations, which are formalized as paths through the HMM.
We had this general idea within the first several months of thinking about the problem together. However, there’s a big difference between writing down an elegant formulation, even one with fast dynamic programming machinery, and actually building a system that scales to data sets of hundreds of thousands or millions of sequences. This is where Duncan showed a tremendous amount of creativity and persistence by assembling layers of approximations and heuristics on top of this essential idea, and by developing a software package meant for others to use.
The code is available as part of the continuing development of partis. It also includes Duncan’s sophisticated BCR simulation package.
We’re under no illusions that we have “solved” this problem, and there’s still a lot to be done. However, we believe that the likelihood-based approach in general and Duncan’s code in particular is a substantial advance over current methods, which use single-linkage clustering based on nucleotide edit distance. If you work with BCR sequences, we hope you’ll give partis a spin and let us know what you think.