When doing computational biology, listen to biologists. I have found them to have remarkable intuition; this can be a gold mine of opportunity for us computational types.
In this particular case, the starting point was the stunningly beautiful work of Gabriel Victora’s lab visualizing germinal center dynamics in living mice. For those not yet initiated into the beauty of B cell repertoire, germinal centers are crucibles of evolution, in which B cells compete in an antigen-binding contest such that the best binder reproduces more. As part of the Victora lab work, they did single-cell extraction and sequencing, which enabled them to quantify the frequency of each B cell genotype without PCR bias or other artifacts. Such single-cell sequencing, and consequent abundance information, is now becoming commonplace. How should we use this abundance information in phylogenetics?
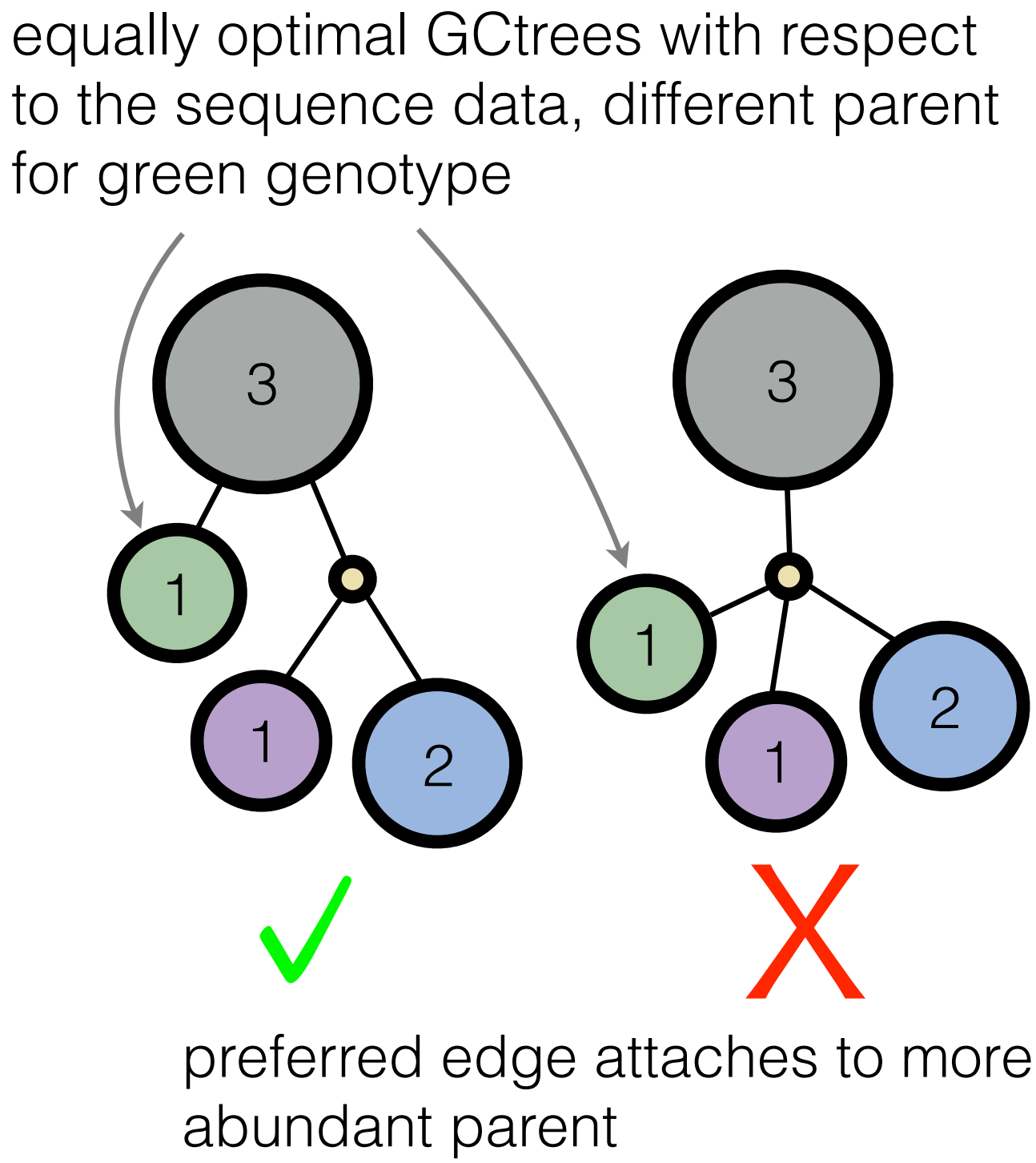
Well, the Victora lab knew, even if their algorithm implementation is not one we would have considered. Indeed, they were building trees by hand, using several criteria about what makes for a believable evolutionary scenario. One of their intuitions was that more abundant genotypes have more opportunity to leave mutant descendants. Therefore, when we are doing inference, we should prefer trees that attach branches to frequently observed genotypes compared to less frequently observed genotypes (see picture, in which the frequency of a given genotype is the number inside the circle; we call this structure a genotype collapsed tree or GCtree).
To have an objective computational method we need to formalize this intuition. Will DeWitt, Vladimir Minin, and I formulated it in terms of an “infinite type” branching process, in which every mutation creates a new type. We can augment existing sequence-based optimality criteria with the likelihood of the tree under our branching process model. In our case we decided to show that this works by ranking maximum-parsimony trees (there are often many equally parsimonious trees). Parsimony is in wide use in the B cell analysis community because it is a defensible choice when sampling is dense relative to mutations (as in the case of germinal centers), and it allows inference of zero branch lengths (leading to inference of sampled ancestral genotypes and multifurcations). We showed under simulation that more highly ranked trees were more correct than lower ranked trees. With the paired heavy and light chain data from the Victora lab, we were also able to do a biological validation by showing that trees that should be the same are more similar when using our algorithm than without. The result is now up on arXiv.
If you are muttering to yourself that we should be using this model as a prior for a Bayesian analysis, we hear you. Hopefully this motivates additional work in that sphere for abundance-based models. We do note that given the limited amount of mutation described before will lead to a fairly flat posterior. Furthermore, although one can infer sampled ancestors using an RJMCMC and multifurcations using phycas, these two features do not exist yet under one roof.
Will did a great job with this project, which is a nice complement to his existing publications as he heads into the UW Genome Sciences PhD program! We had a great time working with Luka and Gabriel, and look forward to more collaboration in the future.