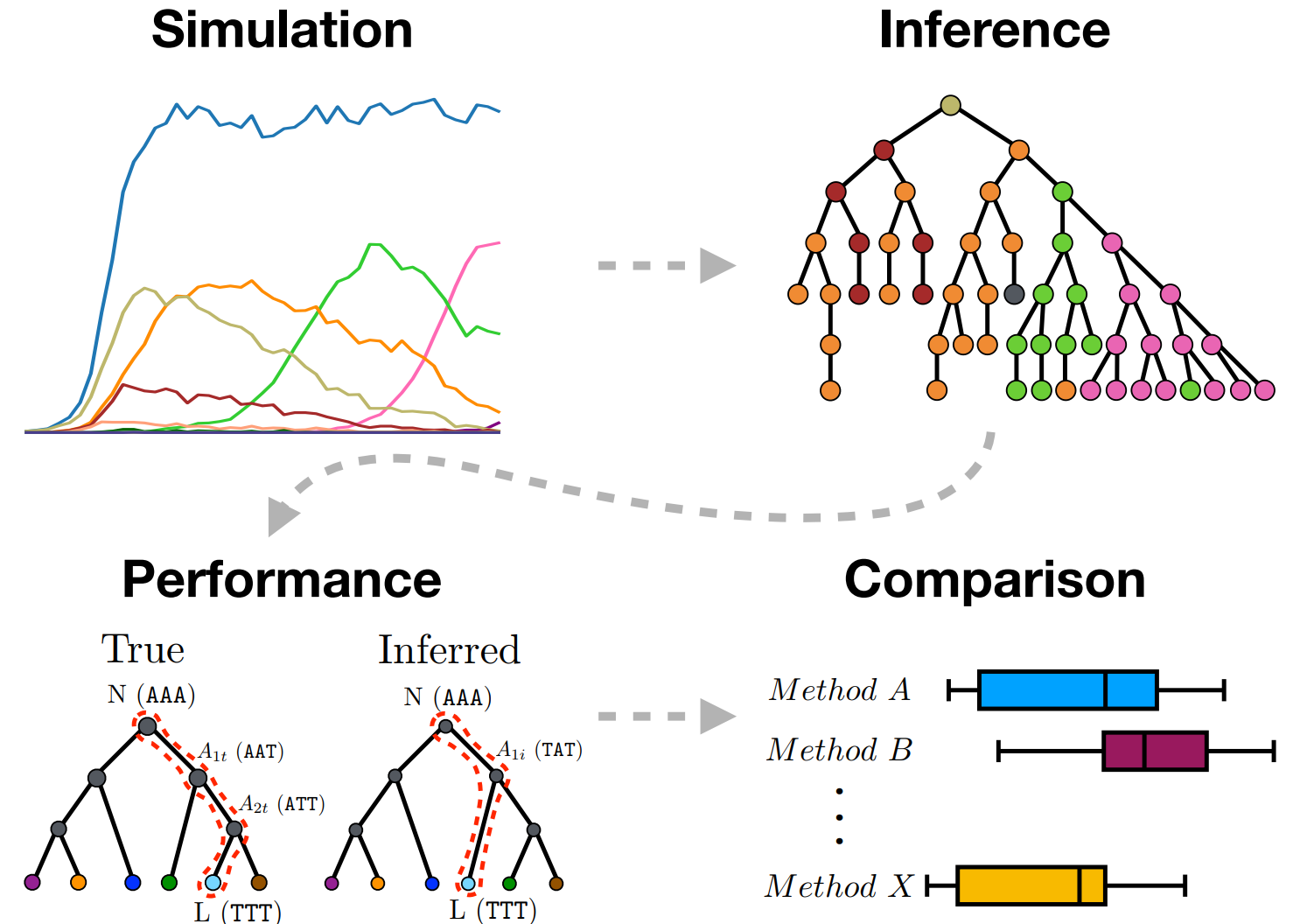
Phylogenetic tools, in particular for ancestral sequence reconstruction, get used a lot in the B cell receptor (BCR) sequence analysis world. For example, they get used to reconstruct intermediate antibodies that then get synthesized in the lab and tested for binding (Wu et. al, 2011). But how well do phylogenetic tools work in this parameter regime? Although there have been countless benchmarking studies for phylogenetics, the case of B cell sequence evolution is different than the usual setting for phylogenetics:
- Sampling and sequencing, especially for direct sequencing of germinal centers, is dense compared to divergence between sequences. Because of the resulting distribution of short branch lengths, zero-length branches and multifurcations representing simultaneous divergence are common.
- The somatic hypermutation (SHM) process in affinity maturation is highly nucleotide-context-dependent process.
- Repertoire sequencing typically focuses on the coding sequence of antibodies, which are under very strong selective constraint. This contrasts with the neutral evolution assumptions of most phylogenetic algorithms, as well as the simulation software assumptions traditionally used for phylogenetics benchmarks.
- In contrast to typical phylogenetic problems where the root sequence is unknown, one has significant information about the root sequence for BCR sequences: namely, that it’s a recombination of V, (D), and J genes, which are somewhat well characterized.
BCR sequences also offer additional opportunities for validation. Specifically, the irreversible class switching process gives us a marker that should only go in one direction along a tree branch. If it goes another direction, this indicates problems with the tree reconstruction.
Before I sketch the results of our analysis, I should mention differences between our work and another recent paper also set up a benchmark of phylogenetic methods. Much of that paper concerns the results of phylogenetic inference using a “toy” clonal family inference method with necessarily bad performance, whereas here we assume that clonal families have been properly inferred. In addition, we simulate sequences under selection using an affinity-based model (which we show makes the inferential problem significantly more difficult), we compare accuracy of ancestral sequence inference, we include additional software tools (several of which are BCR-specific), and we use class-switching data as a further non-simulation means of benchmarking methods.
For this work, Kristian cooked up a simulator for B cell affinity maturation. Although quite a lot of simulators have been written, going back to Clone, none of these did what we wanted, which was to use a context model to simulate mutations, and then use the corresponding amino acid sequences for a selection step. Kristian’s model is simple, but nonetheless we feel that it does an appropriate job of simulating sequences for the purposes of benchmarking methods. We show that the simulated data broadly speaking “looks like” germinal center data.
You can read the full results on bioRxiv, but here are the things that surprised us:
- Picking between equally parsimonious trees using a context-sensitive model works surprisingly well. This makes us want to continue working on incorporating full context models into phylogenetic methods.
- PHYLIP is quite a good choice! I thought that the BCR community was fairly behind the times by not using some of the more modern maximum-likelihood packages, but IQ-TREE is the only recently-developed package that does ML on trees and ancestral sequence inference, and it performs significantly worse (although it’s much faster and nicer to use!).
- IgPhyML is a cool project that works to integrate hotspot motifs and Goldman-Yang codon modeling, which it does by marginalizing out hotspot motifs when they extend across a codon boundary. It does reasonably but not as well as we expected, which may be because we are benchmarking on the moderately-sized trees with which we have experience rather than the very deep broadly-neutralizing trees investigated in the IgPhyML paper.
- The class-switching data gave noisier results than we had hoped for, giving error bars of the same magnitude as differences between methods. However, it confirmed that picking equally parsimonious trees using a context-sensitive model increases accuracy. Perhaps with better sampling or just more data we can learn more from class-switching data in the future.
There’s quite a lot more to do here, both in terms of method development and benchmarking, and we look forward watching this area mature in the coming years. Thanks to Kristian for his great work!