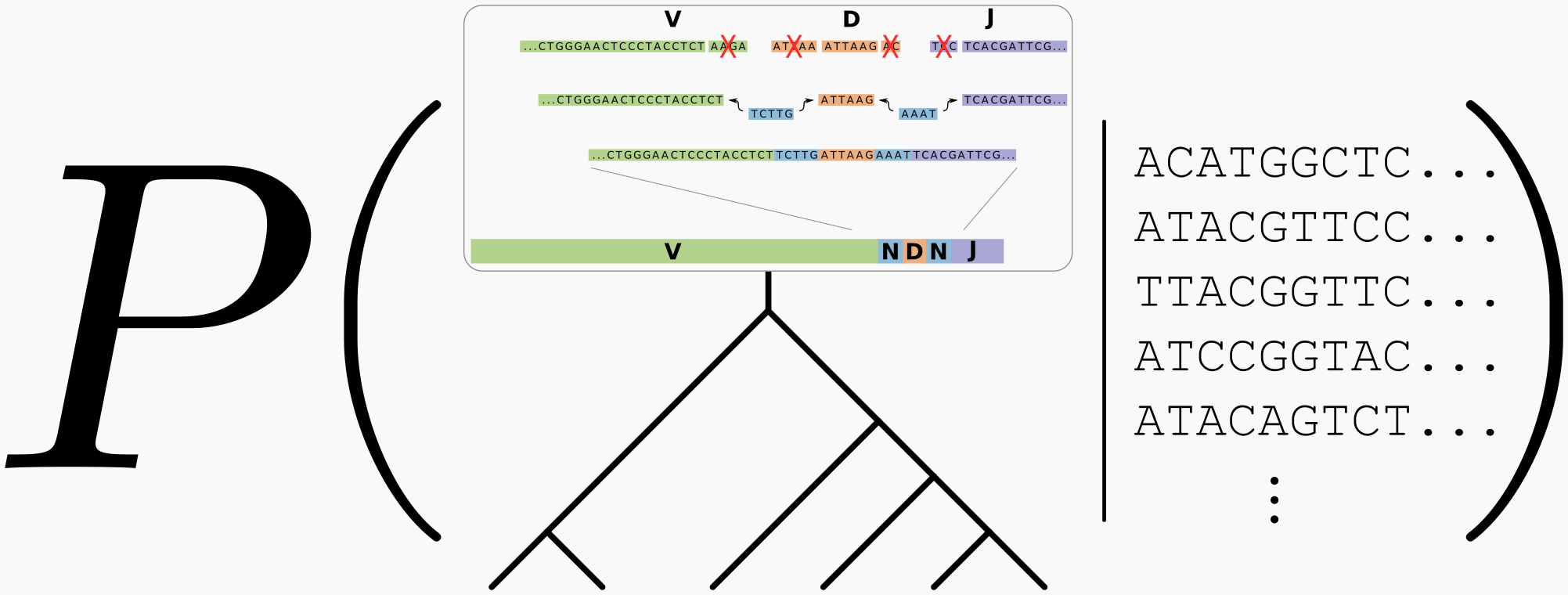
Summary
- antibodies develop within you via an evolutionary process
- understanding these evolutionary patterns is important for understanding how we respond to infection and vaccination
- we have found using Bayesian methods that evolutionary inferences are uncertain in this regime
- our most recent work develops a “Bayesian phylogenetic hidden Markov model,” which takes into account uncertainty in both the V(D)J recombination process and the evolutionary process
- this work reveals substantial amino-acid uncertainty in the inference of the unmutated common ancestor of VRC01, an important and heavily-studied anti-HIV antibody
- our results are described in a preprint which is now being revised for PLOS Computational Biology
A brief description of antibody affinity maturation
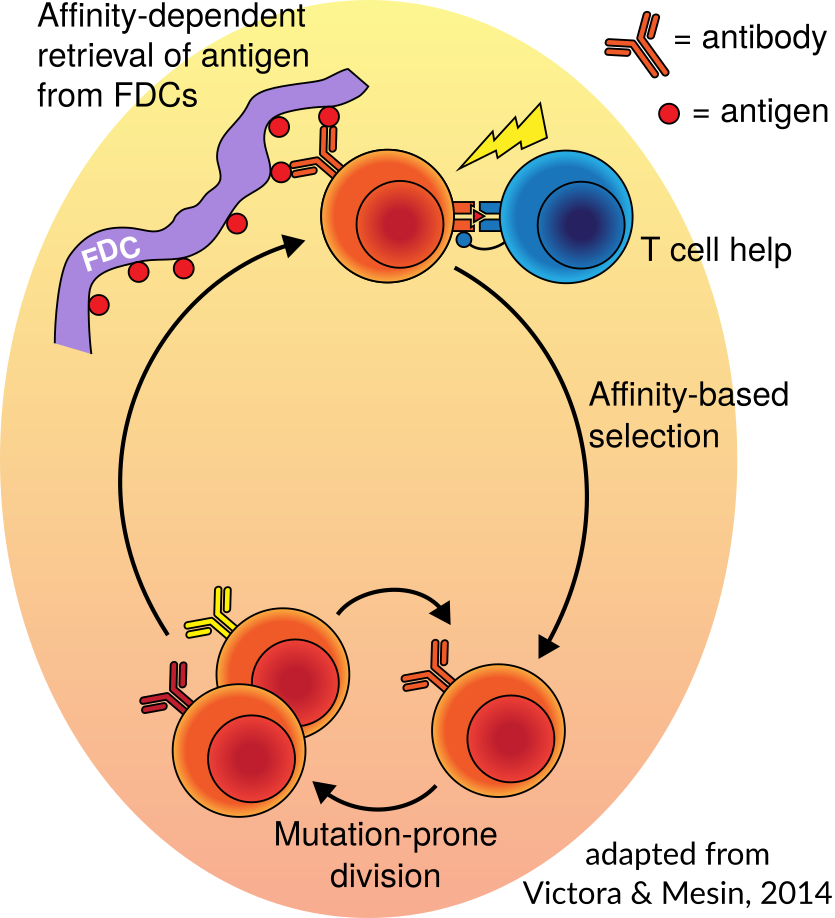
In order to defend against a very large and ever-mutating pool of pathogens, your body randomly generates, and then optimizes, a large collection of antibodies. These antibodies are displayed as so-called B cell receptors on the surface of specialized B cells. The random generation is a process called V(D)J recombination, in which a collection of candidate genes are randomly selected, trimmed a random amount, and then joined by random nucleotides. The optimization is called “affinity maturation,” in which antibody-making B cells are rewarded for being able to bind antigen by being allowed to divide, during which they further mutate their B cell receptor to continue improving binding. . The left hand image below is a cartoon of affinity maturation, simplified from a diagram in Victora and Mesin (2014).
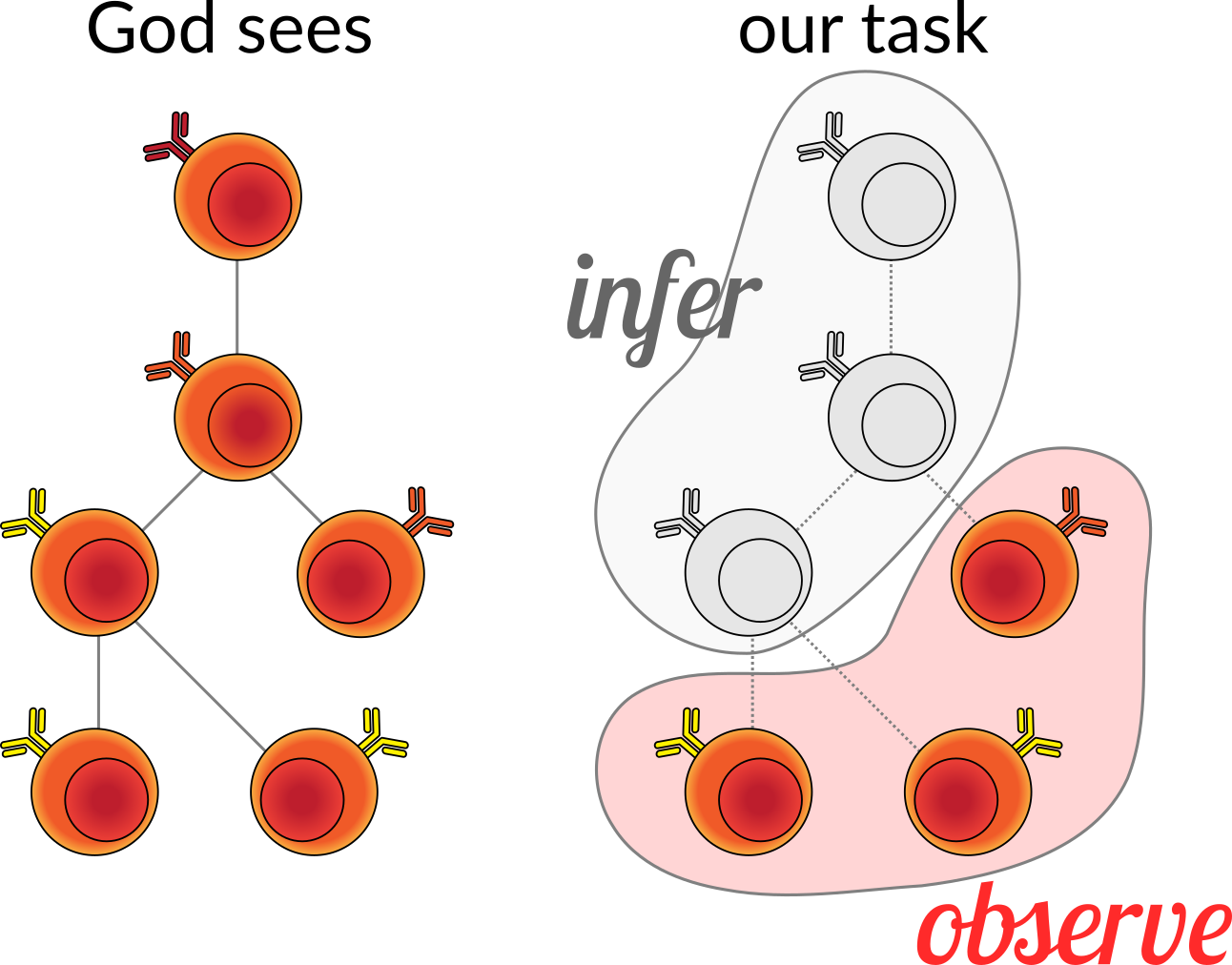
If we were omniscient, we would be able to see this process unfold and record the series of division events and the genetic sequences of the B cell receptors. But, as mere mortals, we have to be satisfied with sequencing some subset of the cells and then reconstructing the process that led to these observed cells. This includes both the tree structure, as well as the states at the internal nodes of the tree. This is a little more complex than the usual phylogenetic tree and ancestral state reconstruction, because we have partial (but highly informative) knowledge about the ancestral state: we know it was sampled from some random process for which we know the ensemble of genes that could have rearranged in order to make the unmutated ancestral sequence. More on this below.
We would like to know the pathways of affinity maturation
The goal of vaccination is to stimulate and affinity-mature antibodies that will be able to block infection. Therefore, if we wish to design better vaccines and understand the impact of existing vaccines, we can use sequencing and sequence analysis methods to understand the development of antibodies. This might be in response to a vaccine, or it might be in response to a viral infection.
Such analysis is especially important for difficult viruses such as HIV. Despite decades of research and an enormous global budget, we still do not have an effective vaccine. This failure stems largely from HIV’s astounding diversity and mutation rate, which make it incredibly difficult for your body to make antibodies that block a usefully large range of HIV strains. Specifically, development of such antibodies typically requires a lot of mutation, including some relatively rare events.
In order to better understand what we can do to stimulate these difficult-to-elicit antibodies, the research community is studying the success stories: individuals that do manage to make antibodies that block a diversity of HIV strains. These super-antibodies are called “broadly neutralizing antibodies” or bNAbs. There is a tremendous amount of interest in a particular bNAb called VRC01. For example, there is currently a clinical trial to find if VRC01 infusion can block infection. Although this trial will tell us if VRC01 can block HIV infection in a realistic setting, what we really want is to be able to immunize such that the body makes something like VRC01 from scratch.
This is why it’s important to get a detailed understanding of the steps of affinity maturation taken by VRC01, which means understanding the unmutated common ancestor, as well as the series of mutations. The better we can understand the history of such antibodies, the better we can understand the barriers to eliciting them with vaccination, and the better we can design vaccinations to overcome those barriers.
Correspondingly, researchers have made beautiful and detailed computational dissections of this lineage and of other antibodies in the same class. A 2018 paper had as a primary result a new inference of the unmutated common ancestor. These computational methods are then followed by biochemical analysis of these predicted sequences, which will guide vaccine design.
These biochemical characterizations are only meaningful to the extent that the computational inferences are correct. Next I will describe the main point of this post, which is that if we use Bayesian methods we find that antibody phylogenetics is uncertain.
Bayesian phylogenetic methods describe tree uncertainty
Given a model of sequence evolution and some sequence data, there is in general some ambiguity about the correct evolutionary history. This is depicted in the following cartoon:
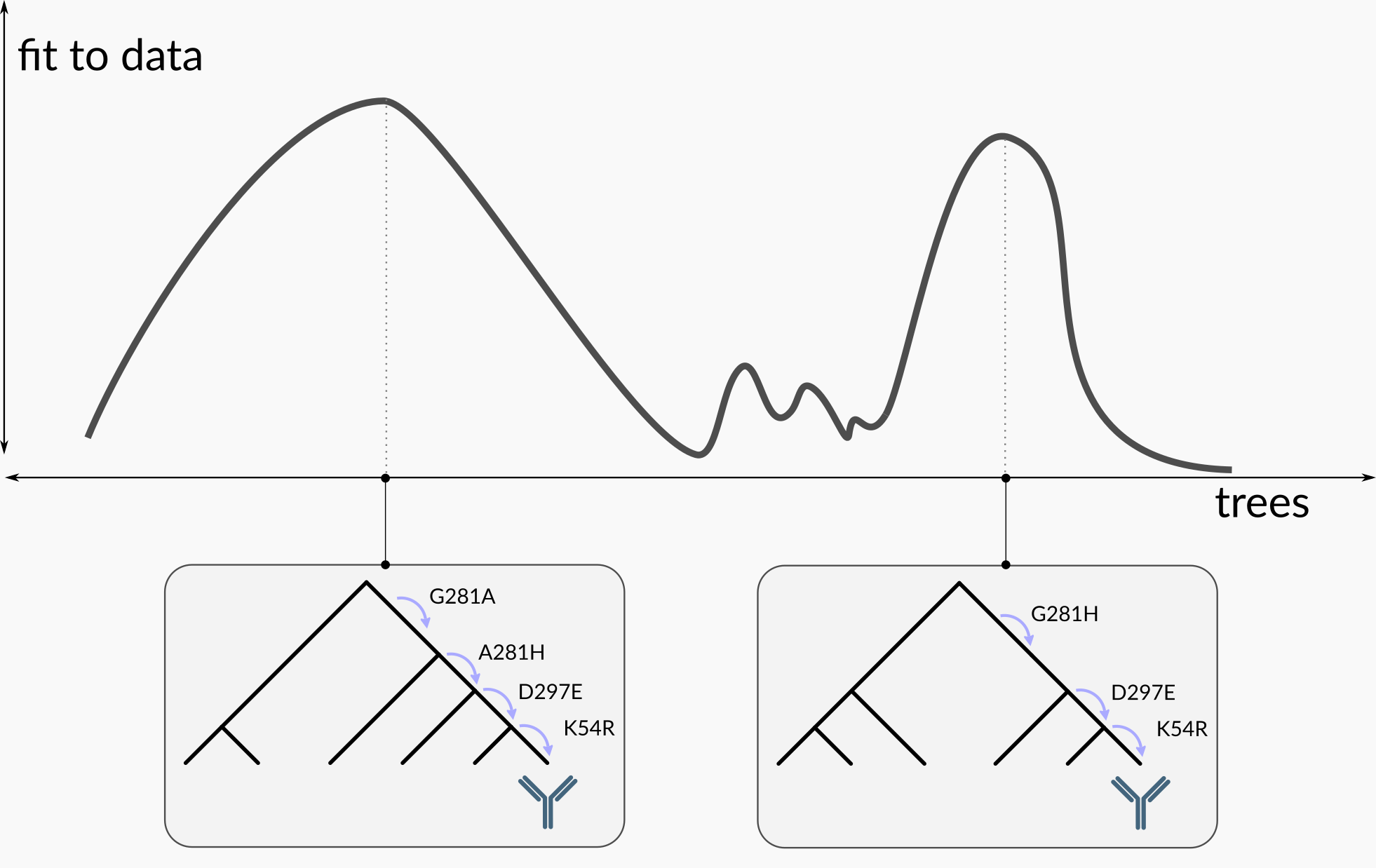
If we are only interested in the best-fitting tree, we can pick the left-hand one, but it fits the data only a little better than the other one. Thus if we really care about the outcome of the analysis, we should consider both of these trees as potential explanations of the data. The goal of Bayesian phylogenetics is to find all of the credible phylogenetic trees, and assign each of these a probability that it is the correct tree.
We can “boil” these trees down to quantities of actual interest to researchers. For example, antibody researchers are commonly interested in the sequence of mutations leading to a specific antibody of interest, rather than the full tree containing those mutations. We can represent the possible mutation paths and their probabilities in a diagram like this one, which was made using a simpler version of the methods described in this blog post. We were inspired in this representation by lovely work from Jesse Bloom’s lab.
When we apply Bayesian methods to real data, we find substantial uncertainty in antibody trees. This wouldn’t surprise an experienced phylogenetics researcher, because antibody sequences are relatively short, and mutation is typically focused in specific areas. Thus, we think that Bayesian methods should be the method of choice for researchers who care a lot about the details of the sequence of mutations leading to an antibody of interest. (If you cared a lot about the slope of a regression, wouldn’t you want to get a confidence interval? 😊)
A Bayesian phylogenetic hidden Markov model for B cell receptor sequences
There is something special and interesting about phylogenetics in this regime: we have information about the sequence at the root of the tree. This is because we know that it came from V(D)J recombination, and there are databases of the various V, D, and J genes present in the population that go into that recombination. We can formalize this knowledge as a probabilistic model of V(D)J recombination, which can be used as a prior on root sequences for our B cell phylogeny.
Putting this together with the usual Bayesian phylogenetic machinery, we have a posterior that looks like so:
where the box at the top of the tree is meant to represent a probabilistic model of the V(D)J recombination process. Samples from this posterior integrate uncertainty in both the recombination process and the phylogenetic tree.
Amrit Dhar, a statistics PhD student working with Vladimir Minin and me, led development of a way of sampling from the posterior of these structures. In doing so, he had to cope with the complexities of V(D)J recombination modeling (with assistance from Duncan Ralph), as well as the complexities of doing Bayesian phylogenetics. The methods and validations are described in a preprint which is now being revised for PLOS Computational Biology.
Substantial uncertainty in the unmutated ancestor of VRC01
Using Amrit’s method, we find substantial uncertainty in the inference of the VRC01 unmutated common ancestor for the CDR3 region, which is a key region determining binding. We can visualize it like so, with the heights of the letters being the posterior probability of that letter’s amino acid at each site:
![]()
Substantial uncertainty is evident at a number of sites, and with quite different amino acids. For example, Tyrosine (Y) and Asparagine (N) have very different biochemical properties. We might expect these variants to have different binding properties.
On the other hand, when the data supports a single clear answer, then the method reports it. For example, we find very little uncertainty in the inferred ancestor of PC64, another antibody lineage of great interest to HIV researchers.
Some final thoughts
Bayesian methods are expensive to run, and it would require a staggering amount of compute to run this method on every inferred clonal family in a repertoire. We don’t. We only run it when we really care about understanding the collection of predicted ancestral sequences, such as when we want to express inferred antibodies and test their properties in the lab.
There’s a lot left to do here. We would like to make the method faster (see our other research on accelerating Bayesian phylogenetic inference) so we can scale the method to more sequences. Our method does not take into account the context-sensitive nature of the B cell receptor mutation process, like IgPhyML does. Also, we’d ideally like to have a method that also incorporates uncertainty concerning which sequences are in the tree, which is not known a priori.
Thank you to Amrit, whose incredible determination pushed this challenging project through to completion, Duncan, for VDJ recombination consults and integration with partis, and to Vladimir, for being an awesome collaborator from the vision to the final details. See the preprint for many other thank-yous, but I’d like to especially credit the Overbaugh lab for keeping us motivated to work on this challenging problem.
I would also like to credit Tom Kepler, whose pioneering work gave the first means of integrating phylogeny and rearrangement inference. His software performed remarkably well in our benchmarks for the task of providing a point estimate of the tree.
Please comment and ask questions here.
We’re always interested in hearing from people interested in our work who might want to come work with us as students or postdocs. Please drop me a line!